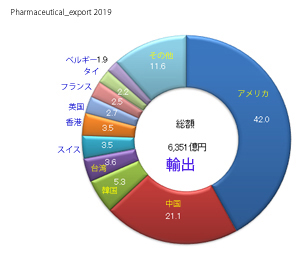
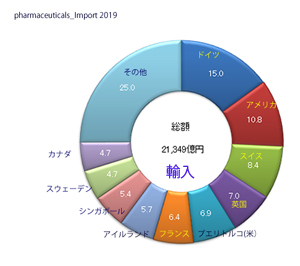
輸入医薬品の管理のための措置
(2019年5月16日に市場規制のための州政府の命令第9号によって公布)2020/1 施行
第1章一般
第1条 輸入医薬品の監督・管理を強化し、輸入医薬品の品質を確保するために、「中華人民共和国薬事行政法」「中華人民共和国薬物行政法施行規則」等の法令等に基づいた措置を講じている。
第2条 これらの措置は、輸入医薬品の申請、審査および承認、提出、港湾検査および監督および管理に適用されるものとする。
第3条 医薬品は、国務院が医薬品の輸入を承認した港または国境港から輸入するものとする。
第4条 国家医薬品局は、全国の輸入医薬品の監督と管理を担当している。州医薬品局は、州、自治区、および地方自治体の医薬品規制機関(以下、州の医薬品規制機関と呼ぶ)に、医薬品の最初の輸入の承認の実施を委託し、医薬品の最初の輸入承認の委託された実施を監督および指導します。
州の医薬品監督管理部門は、法律に従って輸入医薬品を監督および管理し、委託の範囲内で州医薬品局の名義で医薬品の最初の輸入承認を実施するものとします。
医薬品の輸入が許可されている港または国境港の医薬品の輸入が許可されている港の医薬品の監督および管理を担当する部門(以下、港湾医薬品の監督および管理部門)は、輸入された医薬品の記録を担当し、港の検査を組織し、監督および管理を行います。
第5条 これらの措置に記載されている医薬品の輸入事業体とは、医薬品の最初の輸入の承認を取り扱う申請者、または輸入された医薬品の記録を取り扱う事業体を指します。
医薬品の輸入単位は、中国特許医薬品の販売免許の保有者、中国医薬品の製造業者、および中国医薬品または中国医薬品デコレーションピースの事業範囲を持つ製薬企業であるものとします。
第6 条初めて医薬品を輸入する場合は、本措置の規定により輸入医薬品の承認書類を取得した後、港湾医薬品監督管理部に提出してください。医薬品の最初の輸入とは、同じ国(地域)、同じ申請者、または同じ出所からではない輸入された医薬品を指します。
初めての医薬品の輸入については、これらの措置の規定に従い、港湾医薬品監督管理部門に直接提出するものとします。最初に輸入されなかった医薬品はカタログ管理の対象であり、特定のカタログは州の医薬品局によって作成および調整されます。カタログに記載されていないが、申請者、医薬品の拠点、国(地域)が変更されていない場合は、最初に輸入されていない医薬品に基づいて管理するものとします。
第7条 輸入された医薬品は、国の医薬品基準を満たさなければならない。中国薬局方の現行版に含まれていない品種については、輸入医薬品の基準を実施し、中国薬局方の現行版に含まれていない品種および輸入医薬品の基準については、その他の国の医薬品基準を実施するものとします。少数民族地域で慣習的に使用されている少数民族医薬品の輸入に関する国内医薬品基準がない場合、それらは対応する州および自治区の医薬品基準を満たさなければならない。
第II章医薬品の最初の輸入のための申請と承認
第8条:初めて医薬品を輸入する場合、申請者は、州医薬品局の情報システム(以下、情報システム)を通じて輸入医薬品の申請書に記入し、以下の資料を地方の医薬品規制当局に提出しなければならない。
(1)輸入医薬品申請書;
(2)申請者の医薬品製造ライセンスまたは医薬品事業ライセンスのコピー申請者が中国特許医薬品販売許可の保有者である場合、関連する医薬品承認文書のコピーを提供するものとします;
(3)輸出者の主な登録証明書
(4)購入契約書とその公証文書のコピー;
(5)生態環境、資源保護区、野生または植栽および繁殖条件、生産地域での
医薬品の収穫および前処理に関する情報;
(6)医薬品規格および標準ソース;
(7)中国の領土には、資格のある査定に含まれる根拠、医学情報、専門家の結論、サンプル画像、鑑定士、査定機関、および元の身分証明書の元の基盤などの公式シールによって発行された元の植物識別の動的で制度的な基盤があります。
申請者は、申請資料の信憑性に責任を負うものとします。
第9条 医薬品の最初の輸入のための申告資料を受け取った後、州の薬物監督および管理部門は、申告資料の標準化および完全性の正式なレビューを実施するものとする。その場で訂正できる申請資料に誤りがある場合は、その場で訂正することができます。申請資料が不完全であるか、法定書式に適合していない場合は、その場で、または訂正が必要なすべての内容から5日以内に一度に通知するものとします。申し込みは申し込み日から受け付けます。
州の医薬品監督管理部門は、医薬品の最初の輸入の申請を承認または拒否するときに、承認または拒否の通知を発行するものとします。承認されない場合は、理由を書面で説明するものとします。
第10条 申請者は、最初に輸入された医薬品の受理通知を受け取った後、直ちに検査サンプルを所在する州の医薬品検査機関に提出し、同時に本措置の第8条に規定された材料を提出しなければならない。
第11条 地方医薬品検査機関は、試験サンプルおよび関連資料を受け取ってから30日以内にサンプル検査を完了し、輸入医薬品の検査報告書を申請者に発行し、州医薬品監督管理部門に提出するものとする。品種の特性や検査項目により検査期間を延長する必要がある場合は、期限と延長理由を州の医薬品規制当局に書面で報告し、申請者に通知するものとします。
第12条 申請者が検査結果に異議を唱える場合、薬物投与法の規定に従って再検査を申請することができる。薬物検査機関は、再検査申請を受理してから20日以内に再検査の結論を出し、州の薬物規制当局に報告して申請者に通知するものとします。
第13条 審査・承認の過程で、州の医薬品監督管理部門は、申請者が資料を補足する必要があると判断した場合、一度に補足する必要のあるすべての内容を通知するものとする。
申請者は、補足資料の通知を受けてから4か月以内に、必要に応じて1回補足資料を提出するものとします。補足資料が期限内に提出されない場合、不承認の決定がなされるものとします。強制的な威嚇等により、所定の期限内に補足資料を提出できない場合は、延長申請書を地方の医薬品規制当局に提出し、その理由を説明するものとします。
第14条 州の医薬品監督管理部門は、申請の受理日から20日以内に承認または不承認の決定を下すものとする。要件を満たしている人には、輸入された医薬品の1回限りの承認が発行されます。検査および補足資料の期限は、承認の期限には含まれていません。
第15条 輸入医薬品の承認書類の承認項目が変更された場合、申請者は、情報システムを通じて輸入医薬品の補足申請書に記入し、承認文書を最初に発行した州の医薬品規制当局に補足申請書を提出しなければならない。補足申請書の申請者は、原本輸入医薬品承認書の所持者であり、以下の資料を提出しなければならない。
(1)輸入医薬品の補足申請書、
(2)輸入医薬品の原本承認書、
(3)変更に関連する資料。
申請者が名前を変更する場合は、最初の段落で指定された資料に加えて、申請者は、薬剤製造ライセンスまたは薬剤事業ライセンスのコピーと変更記録ページ、または薬剤承認文書と所有者の名前変更の補足申請書も提出するものとします。コピー。
申請者が到着港を変更する場合、最初の段落で指定された資料に加えて、購入契約書のコピーとその公証された文書も提出されなければならない。
第16条 州の医薬品監督管理部門は、補足申請を受けてから20日以内に検査と承認を完了するものとする。要件を満たしている場合は、輸入医薬品の補足申請承認書を発行します。
第17条 州レベルの医薬品規制当局が承認を決定した場合、承認決定がなされてから10日以内に、輸入医薬品承認文書または輸入医薬品補足申請承認文書を申請者に提供するものとする。決定が承認されてから10日以内に、審査意見の通知が申請者に提供され、理由が説明され、申請者は、法律に従って行政再考を申請する権利または行政訴訟を起こす権利を通知されるものとする。
第III章ファイリング
第18条 医薬品の初回輸入申請者は、医薬品輸入承認書を取得してから1年以内に、医薬品輸入承認書に記載された到着港からの医薬品の輸入を手配するものとする。
第19条 輸入事業体は、港湾医薬品監督管理局に提出し、情報システムを通じて輸入医薬品検査用紙に記入し、以下の資料を提出しなければならない。
(1)元の輸入医薬品検査用紙;
(2)原産地証明書の写し;
( 3)医薬品の基準と標準ソース;
(4)パッキングリスト、荷積み請求書、出荷請求書のコピー;
(5)他の国(地域)を通じて再輸出される輸入医薬品は、すべての購入契約を原産地から再輸出地に同時に提出する必要があります、パッキングリスト、梱包請求書、および出荷請求書のコピー;
(6)輸入された医薬品に、絶滅危惧種の野生動植物の国際貿易に関する条約によって制限されている絶滅危惧種の野生動植物が含まれる場合、それらはまた、承認のために国家絶滅危惧種の輸出入管理機関を提供するものとします。インポートおよびエクスポート許可証明書のコピー。
医薬品の初回輸入を申請する場合は、最初の段落で指定された資料に加えて、輸入医薬品承認文書および輸入医薬品補足申請承認文書(ある場合)のコピーも提出するものとします。
医薬品の初回以外の輸入については、最初の段落で指定された材料に加えて、輸入者の医薬品製造ライセンスまたは医薬品事業ライセンスのコピー、輸出者の主要登録証明書のコピー、および購入契約とその公証も提出するものとします。ドキュメントのコピー。輸入部門が中国特許医学の販売免許の保有者である場合、関連する医薬品承認証明書のコピーを提供するものとします。
第20条 港湾医薬品監督管理部門は、申告資料の完全性と標準化について正式な見直しを行い、要件を満たしている場合は、輸入医薬品の通関書類を発行し、輸入医薬品の最初のバッチを撤回すると同時に、輸入医薬品の港を港湾医薬品検査機関に発行するものとする。ファイリング資料のコピーを含む検査通知。
第21条 輸入部門は、輸入医薬品の通関申告書を用いて通関手続きを行うものとする。
第IV章ポート検査
第22条 輸入医薬品の港湾検査通知を受けた後、港湾医薬品検査機関は、現地サンプリング時間に2日以内に輸入業者と交渉し、指定された在庫場所に出向き、現地サンプリングを行うものとする。現場でサンプリングする場合、輸入ユニットは原産地証明書を提示するものとします。
第23条 港湾薬物検査機関は、元の原産地証明書および実際の医薬品の配達と、港湾薬物監督および管理部門から提供された提出資料との整合性を検証するものとする。要件を満たした者をサンプリングし、輸入医薬品サンプリング記録用紙に記入し、輸入ユニットが保有する元の輸入医薬品通関用紙に「サンプリング」と記載し、サンプリングユニットの公式印鑑を押印する。そして、2日以内に地元の港湾薬物監督および管理部門に報告してください。
第24条 港湾医薬品検査機関は、通常、サンプリング後20日以内に検査作業を完了し、輸入医薬品の検査報告書を発行するものとする。客観的な理由により検査が時間通りに完了できない場合、輸入者は期限と延長の理由を書面で通知され、港湾医薬品監督管理部門に報告されなければならない。
港湾医薬品検査機関は、輸入医薬品の検査報告書を港湾医薬品監督管理部門に提出し、輸入部門に通知するものとする。
港での検査に合格した輸入医薬品のみの販売・使用が可能です。
第25条 輸入部門が検査結果に異議を唱えた場合、医薬品管理法の規定に従い、再検査を申請することができる。医薬品検査機関は、再検査申請を受理してから20日以内に再検査の結論を出し、港湾医薬品監督管理部門に報告し、輸入部門に通知するものとします。
第V章監督と管理
第26条 輸入医薬品の非サンプリングの通知を受けた後、港湾医薬品監督管理部門は、人の健康を危険にさらす可能性のある証拠があり、税関検査および解放手続きを経たすべての医薬品を封印および拘留するための行政強制措置を講じるものとする。取り扱いの決定は7日以内に行われます。
第27条 基準要件を満たしていない輸入医薬品について、税関検査・釈放手続きを経た場合、港湾医薬品監督管理部門は、検査報告書受領後、速やかに封印・拘留する行政強制措置を講じ、法令に基づく取り扱い決定を行うものとする。 、同時に、関連する取り扱い状況を地方の医薬品監督管理部門に報告してください。
第28条 国家医薬品局は、必要に応じて、輸入医薬品の生産現場および一次加工現場での海外検査を組織し、実施することができる。医薬品輸入部門は、検査に協力するように輸出業者を調整するものとする。
第29条 輸入医薬品を購入する場合、中国独自の医薬品販売免許の保有者、中国医薬品メーカー、製薬会社は、港湾医薬品検査機関が発行する輸入医薬品検査報告書の写しを確認し、「採取」と記載し、捺印する。輸入医薬品通関用紙の公式シールのコピーは、医薬品の遡及的管理に関連する規制を厳格に実施するものとします。
第30条 輸入医薬品の包装は、輸入医薬品の品質要件を満たし、保管、輸送、輸入検査に便利でなければなりません。各パッケージには、医薬品の中国名、バッチ番号(最初に輸入された医薬品を除く)、原産地、ラベル番号、輸入業者の名前、輸出業者の名前、到着港、重量、および処理と梱包の日付を各パッケージに記載する必要があります。
第31条 医薬品の輸入申請の受理・承認結果、関連する法規制違反、処罰結果は、国家医薬品局のウェブサイトで公表するものとする。
第VI章法的責任
第32条 輸入部門が虚偽の証明書を提出したり、サンプルを文書化したり、その他の欺瞞的手段を用いて医薬品の最初の輸入承認を取得した場合、それは薬物管理法およびその他の法律および規制の規定に従って取り扱われるものとする
。
第33条 輸入部門が記録のために虚偽の証明書、文書またはその他の欺瞞的手段を提供した場合、警告が発せられ、10,000元から30,000元までの罰金が科せられる。
第7章補足規定
第34条 輸入医薬品の承認書の番号形式は、(中央政府直属の州、自治区、市町村の略称)医薬品エントリー+4桁の年番号+4桁のシリアル番号です。
第35条 これらの措置は2020年1月1日に発効するものとする。2005年11月24日に旧州食品医薬品局によって公布された「輸入医薬品の行政措置(試験)」は同時に廃止されるものとする。
以上 |